|
Progressive Ataxia
of Unknown Etiology
......................................................................................................................................................................
Abdulrazak Abyad
Correspondence:
A. Abyad, MD, MPH, MBA, AGSF , AFCHSE
CEO, Abyad Medical Center
Chairman, Middle-East Academy for Medicine of
Aging
Coordinator, Middle-East Primary Care Research
Network
Coordinator, Middle-East Network on Aging
Email: aabyad@cyberia.net.lb
The hereditary ataxias are a
heterogeneous group of diseases. Most attempts
at classification have been based on pathologic
findings and are not always useful for the clinicians.
Many of these disorders are multisystem degeneration
in which the underlying biochemical or other defect
is usually unknown. The pathophysiology is correspondingly
poorly understood. Hereditary ataxia can be divided
into the hereditary congenital ataxia, the ataxia
linked with metabolic disorder, and early onset
ataxia of unknown etiology (1) ( Table 1).

The degenerative cerebellar and
spinocerebellar disorders are a complex group
of diseases, most of which are genetically determined.
Tremendous confusion exists in classifying degenerative
disorders causing ataxia, and there is no universally
accepted system. These disorders can be divided
into two main groups, depending on whether onset
of symptoms is before or after the age of 20 years.
Most of the early onset are autosomal recessive,
and the later onset ones autosomal dominant (2).
Most of these disorders are multisystem degenerations
in which the underlying biochemical or other defect
is usually unknown; the pathophysiology is correspondingly
poorly understood. The differential diagnosis
of ataxia is important since some of them are
treatable if detected early. The discussion will
concentrate on progressive ataxia of unknown etiology.
| PROGRESSIVE
ATAXIA DISORDERS OF UNKNOWN ETIOLOGY |
These can be divided into two
main groups, depending on whether onset of symptoms
is before or after the age of 20 years. Most of
the early onset disorders are autosomal recessive,
and the later onset ones autosomal dominant
(2).
A. Early Onset Cerebellar
Ataxia
Friedreich's ataxia (FA)
Friedreich's ataxia is the most common of the
early onset ataxia. It is one of the best defined
and most common forms of hereditary ataxias of
unknown etiology(1,2). In some large case series
it comprises about 50% of the hereditary ataxia
(2,3). It is transmitted in an autosomal recessive
manner, with occasional sporadic cases, and usually
appearing in childhood or in adolescence but rarely
in old age (4). The disease usually progresses
slowly without remission, affecting both the central
and peripheral nervous system (4,5). The most
frequent first symptom is ataxia of gait, although
occasionally scoliosis or cardiac symptoms precede
definite neurologic symptoms.
The epidemiology of Friedreich's
ataxia is perplexing. The clinical features and
diagnostic criteria were defined by the Quebec
Cooperative Study of Friedreich's Ataxia (QCSFA)
(6) and by Harding (1,2) (Table 2). Both authors
regarded recessive inheritance, progressive ataxia
of limbs and gait and lower limb areflexia as
obligatory criteria. The onset, according to the
QCSFA and Harding (2, 6), should never occur after
the age of 20 years, and always before 25, according
to Harding (2). A recent case was reported in
the literature where symptoms started at a later
stage (7). Both consider extensor plantar response,
pes cavus, scoliosis and cardiomyopathy frequent,
but not essential signs. Dysarthria, decreased
lower limb deep sensation and weakness, obligatory
signs for the QCSFA, are not considered essential
for an early diagnosis by Harding (2). The diagnosis
is made essentially on clinical grounds; CT scan
of the brain may show mild cerebellar atrophy.

The prevalence is known only
for some populations (3, 8-11). The range is from
0.6 to 1.4/100,000 population. The incidence has
been estimated to be approximately 1-
2/100,000 (8,12).
In Southern Italy it ranges from
2.1 to 5.4 x 10-5 (13). Some studies revealed
female preponderance (14,15); other series revealed
that it occurred equally in males and females
(10,16).
Friedreich's ataxia is characterized
by degeneration of the spinocerebellar pathways,
the dorsolateral columns, and the dentate nuclei
(1). There are few changes in the cerebellar cortex
itself (1). The cerebrospinal fluid is usually
normal and the CT scan of the brain is either
normal or shows mild cerebellar atrophy. The primary
clinical signs include ataxia, most marked in
the lower limbs and often accompanied by dysarthria,
nystagmus is usually present in 70% and skeletal-muscle
weakness (17). Optic atrophy and retinal pigmentation
is usually present. Pes cavus and scoliosis almost
always develop (18). Death is usually sudden and
may be secondary to cardiac arrhythmias (17).
Cardiac involvement is frequent occurring in some
50% to 90% of cases (19); most commonly concentric
hypertrophic cardiomyopathy is found (19,20).
A dilated cardiomyopathy has
been noted only rarely (21,22), and congestive
heart failure is considered a late complication
of the disease. There are suggestions that the
increase in catecholamine release may contribute
to the development of hypertrophic cardiomyopathy
(23). Other authors contest this idea (24).
Multiple studies have shown that
the small coronary arteries are abnormal in patients
who have cardiac disease and Friedreich's ataxia
(25,13). The functional significance of this has
been challenged by Hewer (25). Biller, et al.
(13) reported a prevalence of 1.5% of cerebral
infarction in 131 patients. It occurred in half
of the patients who developed atrial fibrillation
or atrial flutter with underlying symptomatic
cardiomyopathy (13). Speech disorder is common
in FA (14).
Some dysarthric symptoms include:
Sudden pitch changes (this was present in our
patient), ataxic staccato, explosive elements,
transient harshness, disturbances of respiratory
and articulatory control, bradylalia, and dysdiadochokinesia
(26,27).
Electrophysiological and pathological
studies suggest that axon degeneration and secondary
demyelination occur in peripheral sensory nerves
(1 5).
Disease progression is a question
open to discussion. It was suggested (28) that
axon loss in the peripheral nerve may increase
with age. In contrast, some believe (29) that
axon loss does not progress during the disease
and that further clinical worsening may result
from progressive impairment of the cerebellar
and corticospinal pathways (29).
Electrophysiological evaluation of FA patients
usually includes determination of motor and sensory
conduction velocities (MCV, SCV) and multimodal
evoked potentials (30). The degeneration of peripheral
sensory and somatosensory pathway is usually measured
by using nerve conduction studies and somatosensory
evoked potential (SEPs) and brain-stem auditory
evoked potentials (BAEPs) and the blink reflex
(30).
Biochemical alterations observed
in this disease include a reduced insulin receptor
activity which leads to an insulin resistance
state and a reduced glucose tolerance in about
40% of patients (31). Several lipid abnormalities
have been noted as well, including a striking
reduction in linoleic acid (21), low cholesterol
levels with a total cholesterol reduction in serum
and in the LDL and HDL fractions are described
(21). At the cellular level, deficiencies in activity
of the pyruvate dehydrogenase complex and alpha
ketoglutarate dehydrogenase complex have been
described (32).
The results of therapeutic trials
in Friedreich's ataxia with a number of drugs,
including choline chloride, lecithine, physostigmine,
y-vinyl aminobutyric acid, 5-hydroxytrptophan
, benserazide and thyrotropin releasing hormone,
have been inconsistent or unconfirmed in terms
of producing functional neurologic improvement
(2). It was found that the level of the dopamine
metabolite, homovanillic acid (HVA) is low in
the cerebrospinal fluid (CSF) of patients with
either Friedreich's ataxia (FA) or olivopontocerebellar
atrophies (31). Amantadine hydrochloride (AH)
is known to stimulate dopamine release (34). The
use of AH in FA and OPCA was recently tested (35).
Both studies revealed an improvement in reaction
time (RT) and movement time (MT). Surgery for
foot deformity and scoliosis may be of benefit
in well selected patients (36). It is essential
to minimize perioperative bed rest. So there is
no treatment known to influence the slowly deteriorating
disease course. In order to minimize disability
and prolong ambulation, strengthening and stretching
exercises and functional retraining including
aerobic endurance exercise are recommended (36).
Early - Onset Cerebellar ataxia
with Retained Tendon Reflexes
The other early onset ataxias
are listed in Table 3. They are usually rare,
with the exception of early onset cerebellar ataxia
with retained reflexes, which occurs at a frequency
about one quarter of that of FA, is often confused
with it, but is genetically distinct. The main
clinical difference is that the tendon reflexes
are normal or brisk in the disorder (23). It is
important to distinguish between these two disorders,
since the prognosis is better in the former, with
patients losing the ability to walk on average
13 years later than in FA. In addition, severe
skeletal deformity, heart disease, and diabetes
do not occur (24).
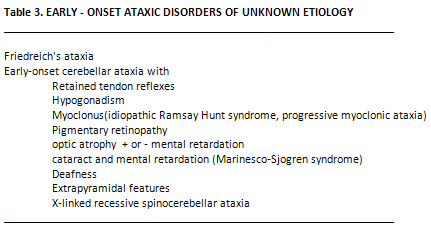
Cerebellar Ataxia with Hypogonadism
The association of progressive
ataxia with hypogonadotrophic hypogonadism is
rare (2). Neurological symptoms usually develop
in the third decade and hypogonadism is obvious
at puberty. Neurological syndromes include dysarthria,
nystagmus, progressive limb and gait ataxia, mental
retardation, dementia deafness, choreoathetosis,
retinopathy and sensory loss.
Cerebellar Ataxia with myoclonus
The association of cerebellar
ataxia and myoclonus, is often referred to as
the Ramsay Hunt syndrome. This is a very heterogenous
entity. Some of the identifiable causes include
Baltic myoclonus, mitochondrial encephalomyopathy,
and sialidosis (24). The rest of cases can be
labelled as progressive myoclonic ataxia (24).
Symptoms include the development of stimulus -
sensitive myoclonus or generalized seizures at
the end of the first decade of life. Ataxia and
dysarthria develop a few years later with pyramidal
signs in the limb. The myoclonic part of this
syndrome may respond to clonazepam or valproate
sodium with marked improvement in motor function.
B. Late Onset
Cerebellar Ataxia
These disorders have proved the
most difficult and controversial in terms of classification
(Table 4). The pathological findings are heterogenous
reflecting huge clinical variations in the dominant
ataxia (2).

Autosomal Dominant Cerebellar
Ataxia Type I (ADCA Type I).
The age of onset of symptoms
in this syndrome ranges from 15 to 65 years but
is most commonly in the third or fourth decade
of life. Ataxia of gait is the most frequent presenting
symptom;, it usually involves the limbs and is
invariably associated with dysarthria. Early onset
usually predicts more progressive disability (37).
Associated symptoms may include ophthalmoplegia,
nystagmus, lid retraction and optic atrophy. Bulbar
symptoms are common during the later stages of
disorder and predispose the patient to respiratory
infection. Other common symptoms include dementia,
extrapyramidal signs, wasting and fasciculation
of the face and tongue.
Autosomal dominant cerebellar ataxia Type II (ADCA
Type II).
This is clinically and genetically
different from ADCA type I. It is characterized
in all families having retinopathy. The age at
onset is earlier than that of ADCA type I, most
commonly occurring between 15 and 35 (2,38).
Autosomal Dominant Cerebellar
Ataxia Type III
This is relatively pure
cerebellar syndrome in which dementia, ocular
or extrapyramidal features do not occur and onset
of symptoms usually after the age of 50 years(39).
Nystagmus and pyramidal signs in the limbs are
quite common.
1. Harding AE Friedreich's
ataxia: a clinical and genetic study of 90 families
with an analysis of early diagnostic criteria
and intrafamilial clustering of clinical features.
Brain 104:589-620, 1981b.
2. Harding AE (1984) The hereditary ataxias and
related disorders. Churchill Livingstone, Edinburgh,
pp 57-103.
3. Berginer V.M., Salen G., Shefer S. Long term
treatment of cerebrotendinous xanthomatosis with
chenodeoxycholic acid. N. Engl. J. Med. 311: 1649-1651,
1984.
4. Petty R.K.H., Harding A.E., Morgan-Hughes J.A.
The clinical features of mitochondrial myopathy.
Brain 109: 9156-938, 1986.
5. Gatti RA, Swift M . Ataxia telangiectasia.
Genetics, neuropathology , and immunology of degenerative
disease of childhood. New York: Alan R Liss, 1985
6. Cleaver J.E. Xeroderma Pigmentosum. In Stanbury
J.B., Wyngaarden J.B., Freidrickson DS, Goldstein
JL , Brown MS, eds. The metabolic basis of inherited
disease. 5th ed. New York: Mc-Graw-Hill; 1983
(57): 1227-50
7. Sjogren T (1943) Klinische and Erbbiologische
Untersuchungen uber die Heredoataxien. Acta Psychiatr
Neurol Scand 27 (suppl):1-200.
8. Stumph DA (1985) The inherited ataxias. Neurol
Clin, 2:47-57.
9. Barbeau A (1982) A tentative classification
of recessively inherited ataxias. Can J Neurol
Sci 9:95-98.
10. Chamberlain S, Shaw J, Wallis J, Rowland A,
Chow L, Farral M, Keats B, et al (1989) Genetic
homogeneity at the Friedreich ataxia locus on
chromosome 9. Am J Hum Genet 44:518-521.
11. Abyad.A, Kligman. E. Hereditary ataxia in
the elderly 1995. J. of International Medical
Research. 23; 1: 74-84.
12. De Michele G, Filla A, Barbieri F, Perretti
A, Santoro L, Trombetta L, Santorelli F, Campanella
G (1989) Late onset recessive ataxia with Friedreich's
disease phenotype. Journal of Neurology, Neurosurgery,
and Psychiatry 52:1398-1401.
13. Romeo G, Menozzi P, Ferlini A, Fadda S, Di
Donato S, Uziel G, Lucci B, Capodaglio L, Filla
A, Campanella G (1983) Incidence of Friedreich's
ataxia in Italy estimated from consanguineous
marriages. Am J Hum Genet 35:523-529.
14. Dunn HG (1973) Nerve conduction studies in
children with Friedreich's ataxia and ataxia telangiectasia.
Dev Med Child Neurol 15:324-337.
15. Tyrer JH (1975) Friedreich's ataxia. In Vinken
PJ, Bruyn GW (eds) Handbook of Clinical Neurology,
Vol 21. Amsterdam, North Holland pp 319-364.
16. Harding A.E. The clinical features an classification
of the late onset autosomal dominant cerebellar
ataxia: a study of elven families including descendants
of the " Drew family of Walworth" Brain
105: 1-28, 1982
17. Polo JM, Calleja J, Combarros O, Berciano
J (1991) Hereditary ataxias and paraplegias in
Cantabria, Spain. An epidemiological and clinical
study. Brain Apr;114(Pt 2):855-66.
18. Martin EA, McLaughlin M, Clarke R, Graham
I, McInerney D, Dean G (1988) Friedreich's ataxia
in Ireland; a clinical and investigative study.
Journal of Neurology, Neurosurgery and Psychiatry,
51, 1363.
19. De Michele G, Filla A, Barbieri F, Perretti
A, Santoro L, Trombetta L, Santorelli F, Campanella
G (1989) Late onset recessive ataxia with Friedreich's
disease phenotype. J Neurol Neurosurg Psychiatry
52:1398-1401.
20. Barbeau A (1980) Distribution of ataxia in
Quebec. In I Sobue (ed): "Spinocerebellar
Degeneration." Tokyo: Univ Tokyo Press, pp
121-142.
21. Perloff JK (1972) Cardiac involvement in heredofamilial
neuromyopathic diseases. Cardiovasc Clin 4:333.
22. Ruschhaupt DG, Thilenius OG, Cassels DE (1972)
Friedreich's ataxia associated with idiopathic
hypertrophic subaortic stenosis. Am Heart J 84:95.
23. Pasternac A, Wagniart P, Olivenstein R et
al (1982) Increased plasma catecholamines in patients
with Friedreich's ataxia. Can J Neurol Sci 9:195-203.
24. Nadas AS, Alimurung MM, Sieracki LA (1951)
Cardiac manifestations of Friedreich's ataxia.
N Engl J Med 244:239-44.
25. Barbeau A, Roy M, Sadibelouiz M, Wilensky
MA (1984) Recessive ataxia in Acadians and "Cajuns".
Can J Neurol Sci 11:526-544.
26. Murray T (1983) Friedreich's ataxia. In W.
Perkins (ed) Current therapy of communication
disorders-dysarthria and apraxia. Thieme-Stratton,
New York.
27. Gilman S, Kluin K (1985) Perceptual analysis
of speech disorders in Friedreich disease and
olivopontocerebellar atrophy. In JR Bloedel, J
Dichgans, & W Precht (eds) Cerebellar functions.
Berlin/Heidelberg: Springer-Verlag.
28. Ouvrier RA, McLeod JG, Cochin TE (1982) Friedreich's
ataxia: Early detection and progression of peripheral
nerve abnormalities. J Neurol Sci 55:137-145.
29. Dyck PJ, Lais AC (1972) Evidence for segmental
demyelineation secondary to axonal degeneration
in Friedreich's ataxia in Kakulas BA (ed) Clinical
studies in Miology. Amsterdam, Excerpta Medica
pp 253-263.
30. Bell CF, Kelly Jm, Jones RS (1986) Anaesthesia
for Friedreich's ataxia. Case report and review
of the literature. Anaesthesia Mar;41(3):296-301.
31. Walton JM (1977) Brain's disease of the nervous
system, 8th ed. Oxford University press 672-4.
32. Blass JP, Sheu RK-F, Cedarbaum JM (1988) Energy
metabolism in disorders of the nervous system.
Rev Neurol (Paris) 144-543-563.
33. Botez MI, Young SN, Botez T, et al (1989)
Treatment of Friedreich's ataxia with amantadine.
Neurology 39:749-750 (letter).
34. Gottdiener JS, Hawley RJ, Maron BJ, Bertorini
TF, Engle WK (1982) Characteristics of the cardiac
hypertrophy in Friedreich's ataxia. Am Heart J
103:525.
35. St. John Sutton MG, Olukotun AY, Tajik AJ,
Lovett JL, Biuliani ER (1980) Left ventricular
function in Friedreich's ataxia. An echocardiographic
study. Br Heart J 44:309.
36. Shapiro F., Bresnan M.J. Orthopaedic management
of childhood neuromuscular disease. II. Peripheral
neuropathies, Friedreich's ataxia and arthrogryposis
multiplex congenita. J. Bone Joint Surg. 64A:949-953,
1882
37. Hewer RL (1969) The heart in Friedreich's
ataxia. Br Heart J 31:5-14.
38. Lamarche JB, Cote M, Lemieux B (1980) The
cardiomyopathy of Friedreich's ataxia: morphological
observations in 3 cases. Can J Neurol Sci 7:389-96
|
